It's better to create a new python environment because I found condaThe default installation of blast is python==3.6.11, you may accidentally change your python version...and then all the code you wrote will die...
conda create -n blast python==3.6.11source activate blastconda install -c bioconda blastnr and uniprot are more general databases:
ftp://ftp.ncbi.nlm.nih.gov/blast/db/
https://www.uniprot.org/downloads
1) nr is the protein sequence of all current microorganisms collected by ncbi, which is used to calculate the frequency of amino acids in general, 160G
2) uniprot90 does a de-redundancy based on similarity, so it is much smaller than nr, 56G
# Take uniprot90 as an examplewget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz # downloadgzip -d uniref90.fasta.gz # unzipmakeblastdb -in uniref90.fasta -parse_seqids -hash_index -dbtype prot # CompileWhat it looks like after parsing:
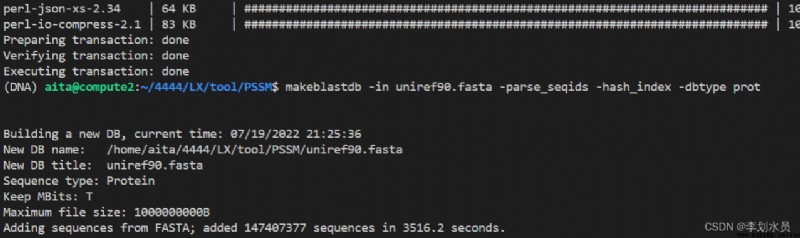
The file looks like this: (only a part of it is intercepted)
My initial file is:
P00269.fasta is for single protein processing, the format inside is:

testset.fasta is a batch process for protein collections. The format inside is (you can also save individual proteins as .fasta files. Since blast can only process a single protein paste, the meaning of summarizing the knowledge of this collection is the first step.To generate a single protein .fasta file, so this file depends on personal wishes):
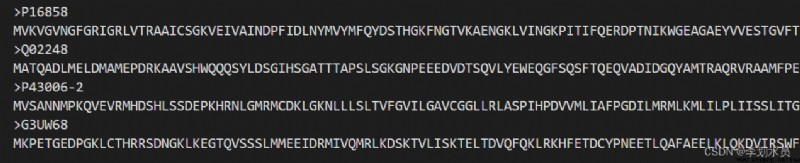
import osos.system('psiblast -query dataset/P00269.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/P00269.pssm')##This protein is so slowimport osfile_name='/dataset/testset.fasta'Protein_id=[]with open(file_name,'r') as fp:i=0for line in fp:if i%2==0:# Protein_id.append(line[1:-1])id=line[0:-1]p=line[1:-1]with open ('/dataset/'+str(p)+'.fasta','a') as protein:protein.write(id)# protein.write()if i%2==1:seq=line[0:-1]with open ('/dataset/'+str(p)+'.fasta','a') as protein:protein.write('\n')protein.write(seq)i=i+1os.system('psiblast -query '+'/dataset/'+str(p)+'.fasta -db /PSSM/uniref90.fasta -num_iterations 3 -out_ascii_pssm /dataset/'+str(p)+'.pssm')##PSSM is really slow, the following is a screenshot after only one is generated
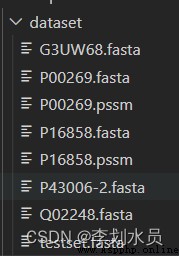
Emmmm, I'm researching how to save this matrix into a file for easy recall. It should be updated today... But it's so slow, I don't want to use it anymore.
References:
Use psiblast under linuxBatch generation of pssm matrix - Chu Junyi's blog - CSDN blog