Python Script editing
Documents used
Input sys modular
Get the file name from the command line
A function for statistics of sequence information
Use def Make a function
.format Use :
Calculate the function
The results are displayed on the screen
Result output file
Script run
Python Script editingUse Python Yes fasta Format sequence for basic information statistics
Expected design output documents include fasta file name , Sequence length ,GC Content and ATCG Respective content .
Documents usedInput sys modulartest.fasta
stat.py
#!/usr/bin/env pythonimport sys Get the file name from the command line file_fasta = sys.argv[1]# Get the filename file_name = file_fasta.split('.')name = file_name[0]sys.argv[1] Module is a bridge to obtain parameters from outside the program , You can enter the parameters of the command line into py In process .
sys.argv[0] It's the program itself ,sys.argv[1] Is the program followed by the first parameter .
We take the file name as the input parameter , This step is shown in the final run .
At the end of the output, a... Containing statistical information will be output txt file , We will use fasta File name as txt File name prefix , So we need to get fasta Name of file .
.split('.') Yes, it will file_fasta With . Separating the separator will generate 'test','txt', Assign a value to file_name be file_name It will contain two characters .
file_name[0] Is the first value 'test',python The default first number in is 0, So you can't type 1.
A function for statistics of sequence informationThe length of the sequence is good for statistical use len Function , however GC Content and ACTG It takes a little work to calculate the percentage of .
Use def Make a functionPython Use def Start function definition , Next comes the function name , Inside the parentheses are the parameters of the function , Internal function Specific function implementation code
def get_info(chr): chr = chr.upper() count_g = chr.count('G') count_c = chr.count('C') count_a = chr.count('A') count_t = chr.count('T')Name this function get_info, The internal parameter is chr
We will fasta in ATCG The base content of is assigned to chr, The base may be uppercase or lowercase , So we use .upper Turn all characters into uppercase .
Reuse .count('G') Statistics ATCG The respective quantities are assigned to the corresponding count_g, We use it ATCG The respective statistics can be immune in the later calculation N Value interference .
gc = (count_g + count_c) / (count_a + count_t + count_c + count_g)A = (count_a) / (count_a + count_t + count_c + count_g)T = (count_t) / (count_a + count_t + count_c + count_g)C = (count_c) / (count_a + count_t + count_c + count_g)G = (count_g) / (count_a + count_t + count_c + count_g)gc_con = '{:.2%}'.format(gc)A_content = '{:.2%}'.format(A)T_content = '{:.2%}'.format(T)C_content = '{:.2%}'.format(C)G_content = '{:.2%}'.format(G)return (gc_con,A_content,T_content,C_content,G_content)gc The content is calculated to be equal to (G The number of +C The number of )/(A The number of +T The number of +C The number of +G The number of )
A The content of is equal to (A The number of )/(A The number of +T The number of +C The number of +G The number of ), Other values are calculated by analogy .
.format Use :Calculate the function"{1} {0} {1}".format("hello", "world") Set the specified location .
'world hello world'
{:.2f} Keep two decimal places
Last , Use return Returns the result of the function (gc_con,A_content,T_content,C_content,G_content)
# Calculate the function with open(file_fasta,'r') as read_fa:Read the contents of the file and assign it to read_fa
python There are two ways to open a file. One is to use it directly open("test.fasta","r"), After execution f.close() close .
notes :"r" Open file in read-only mode ;"w" Open the file in write only mode , In this mode, the input content will overwrite the original content ;"a" Open a file in append mode , This mode will append the new content to the end of the original content , Will not cover .
The second method is used here with Built in functions , It can automatically close the file .
for val in read_fa: val = val.strip() if not val.startswith(">"): seq_info = get_info(val) len_fasta = len(val)take read_fa The content is assigned to val.
strip() Method is used to remove the characters specified at the beginning and end of a string ( The default is space or newline ), The default is used here .
And then use startswith() Method to check whether a string starts with a specified substring , When not > At the beginning of the line , Only then do information statistics on nucleic acid sequences .
len() Method returns the character length to get the fragment length
The results are displayed on the screen# The results are displayed on the screen print('******\n{0}\nlength:{1}\ngc content :{2}\nA content :{3}\nT content :{4}\nC content :{5}\nG content :{6}\n******'.format(name,len_fasta,seq_info[0],seq_info[1],seq_info[2],seq_info[3],seq_info[4]))Use \n Line break , use .format Specify the value output location .
Result output fileos.write(fd, str)
write() Method is used to write a string to a file descriptor fd
# Result output file file_output = open("{}sum.txt".format(name),'a')file_output.write('******\n')file_output.write('{}\n'.format(name))file_output.write('length:{:d}\n'.format(len_fasta))file_output.write('gc content :{}\n'.format(seq_info[0]))file_output.write('A content :{}\n'.format(seq_info[1]))file_output.write('T content :{}\n'.format(seq_info[2]))file_output.write('C content :{}\n'.format(seq_info[3]))file_output.write('G content :{}\n'.format(seq_info[4]))file_output.write('******')file_output.close() Script run Execute the script (linux System )
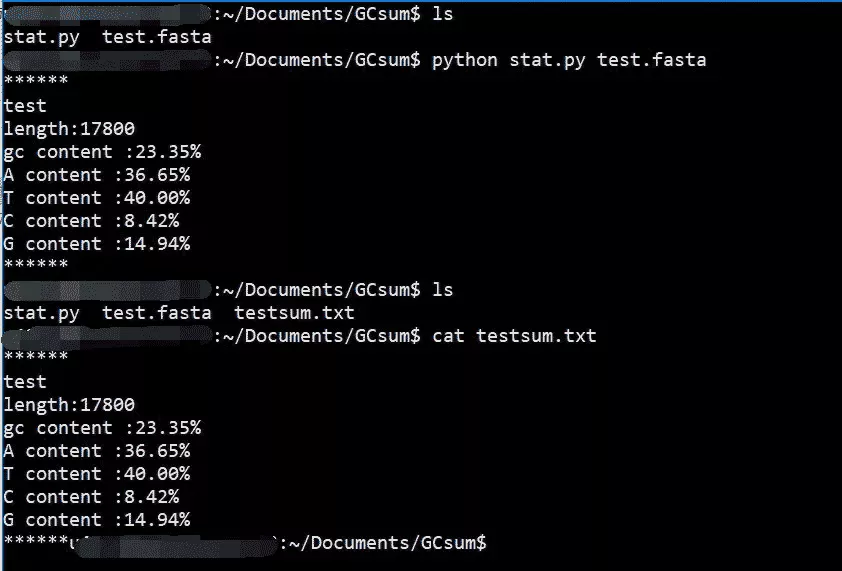
Use ls The command can see that there are already written in the current directory py Documents and data test.fasta.
Note at run time that we set the file name from the command line when writing , So keep up fasta file , Only in this way can it run successfully .
After running, you can see the result print on the screen , It also generates testsum.txt.
Use cat Command view can see the results .
That's all Python Script extraction fasta Details of the implementation of file list sequence information , More about Python extract fasta For single sequence information, please pay attention to other relevant articles on software development network !